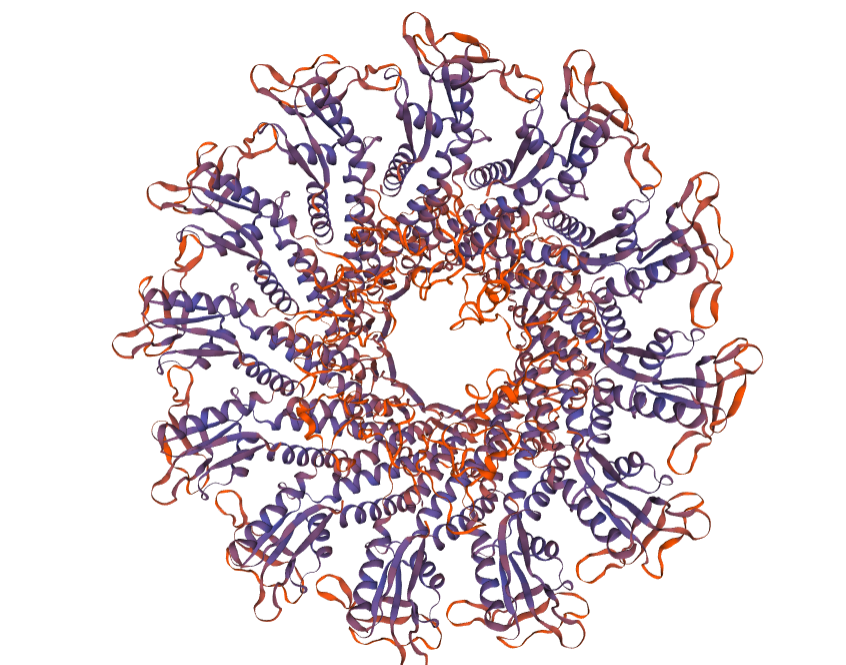
Project at the Arsuaga-Vazquez lab to analyze the connector protein from a database of viruses sequenced from the human gut.
(Above image is the connector complex)
Supporting Code can be found here.
Motivation / Goals
Phages are the most abundant biological entity on Earth and has been shown to have major impacts on human gut health and consequently, human health. But viruses are also known to evolve at rates unheard of in living organisms and this can complicate efforts to draw accurate phylogenetic trees based on sequence alignment as is the usual practice in evolutionary studies. In this project, we attempt to create a more accurate phylogenetic tree based on structural similarities of a key, conserved protein: the connector protein.
Exploratory Data Analysis
The data was obtained from the paper, Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome and contains many (previously) unknown viruses in the microbiome. The majority of the phages in the database are of the order Caudovirales and the 3 major families of this order are shown below:

Workflow
- Data Collection:
- Protein sequences in the MGV database have been clustered into viral protein clusters (VPCs) using MMseqs2 and have been annotated based on various databases such as Pfam, KEGG, and InterPro.
- Extract VPC’s that are annotated with the term “connector” from the MGV database
- Extract protein sequences that belong to these VPCs from the MGV database
- Files for this are located in the scripts directory
- Modelling with AlphaFold2
- We used the AlphaFold2 algorithm to predict the 3D structure of the connector protein sequences extracted as shown above.
- Analysis
- Used TM Align to do pair-wise structural comparisons of the predicted structures + comparisons with known structures of the connector protein (PDB database)
- Structural Analysis
- Structural Phylogenetic tree can be found in tree
- Compare Structural Phylogenetic Tree with Hosts
- hosts can be found in heatmap
Results
It is difficult to verify whether the structural phylogenetic tree we created is any more or less accurate than phylogenetic trees made using sequence alignment. What we do see is that there are certain clades in the structural tree that correspond to certain hosts. This may indicate that the certain hosts are placing some selective pressure on the evolution of the connector protein and the workflow we’ve outlined in this project could potentially used to further explore how hosts or perhaps even other environmental factors influence the evolution of certain proteins.
Future Directions
Instead of using TM-align (which looks at global similarities), we should compare structures based on local similarities. One software that does this is DALI
We only looked compared the structure of the monomer in this project. Using the complete structure of the complex would be more biologically accurate.
And last but not least, it’s always better to have more data. We only used structures in the database that were annotated as “connector”. “Portal” is synonymous with “connector” in bacteriophage physiology. Those structures can also be used here.
Poster presented at URC 2024: